
About
Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes.
Kevin W. Kelley, Hiromi Inoue, Anna V. Molofsky, Michael C. Oldham
Nat Neurosci. 2018 Sep;21(9):1171-1184. doi: 10.1038/s41593-018-0216-z. Epub 2018 Aug 28.
Link to full publication: https://www.nature.com/articles/s41593-018-0216-z
See also News and Views: https://www.nature.com/articles/s41593-018-0218-x
Background
Understanding the molecular basis of cellular identity in the human central nervous system (CNS) is essential for ascertaining the functions of different cell types and their susceptibilities to diverse pathologies. Because gene expression lies at the root of cellular identity, human CNS transcriptomes provide a natural point of entry for this task. However, nearly all gene expression studies of the human CNS have analyzed heterogeneous (bulk) tissue samples comprised of many different cell types. Therefore, it is often assumed that the cellular origins of gene expression in these samples cannot be determined, and that doing so requires physically separating cell types. Although such ‘bottom-up’ methods are readily applied to model organisms, they are difficult to apply to the adult human CNS due to its size, limited accessibility, and resistance to dissociation.
An alternative approach is to estimate the covariation between the abundance of individual cell types and transcripts through integrative gene coexpression analysis of bulk tissue samples. This ‘top-down’ approach assumes that variation in cellular composition among biological replicate samples will drive covariation of transcripts that are uniquely or predominantly expressed in specific cell types. In contrast to single-cell methods, this approach is based on aggregate analysis of many billions of cells, and therefore permits highly robust inferences about the core transcriptional identities of major cell types.
By analyzing gene coexpression relationships in 62 datasets consisting of >7000 neurotypical adult human samples representing all major CNS regions and technology platforms, we identified consensus transcriptional signatures of astrocytes, oligodendrocytes, microglia, and neurons. We created a novel metric called ‘fidelity’, which quantifies the extent to which a gene’s expression levels are correlated with the inferred abundance of a cell type over all analyzed samples. Genes with the highest expression fidelity for a cell type are expressed with high sensitivity and specificity (Fig. 1).
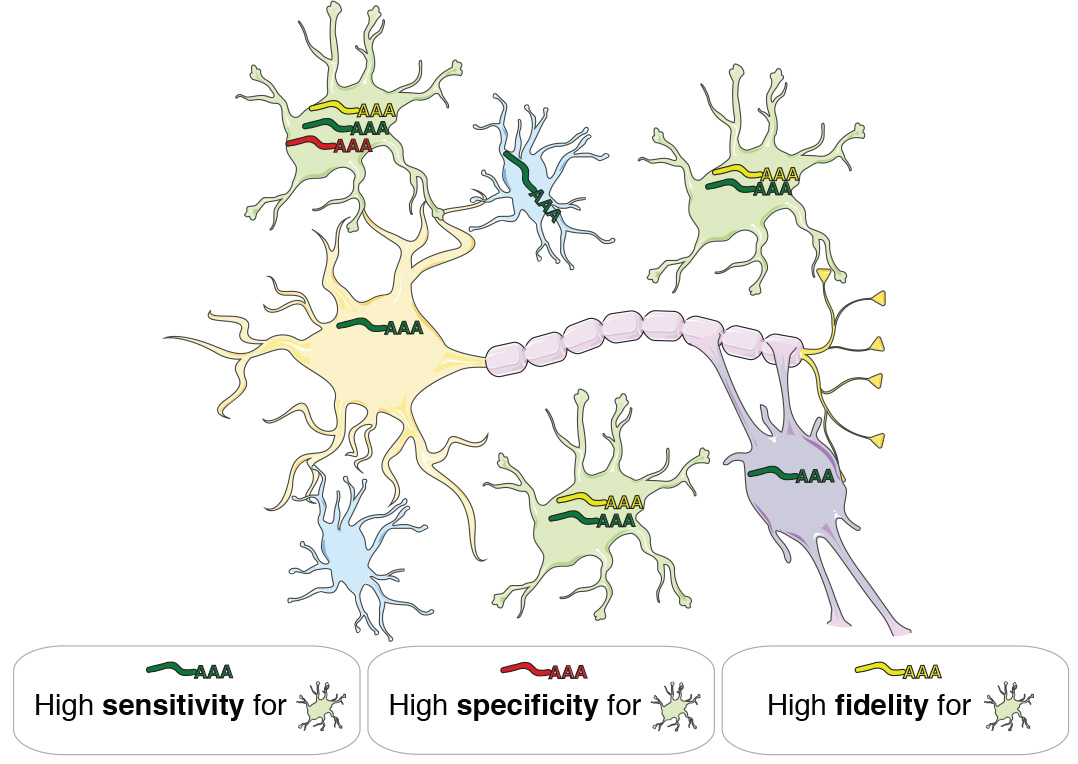
Fig. 1 | Gene expression fidelity. A gene has high fidelity for a cell type if its expression is sensitive (it is consistently expressed by members of that cell type) and specific (it is not expressed by other cell types). Expression levels of high-fidelity genes are therefore highly correlated with the abundance of distinct cell types in heterogeneous tissue samples.
Web site functionality
This web site allows users to explore the covariation of gene expression levels and cellular abundance in the human CNS in two ways:
- CNS region / cell type search: The user can search by CNS region / cell type to retrieve the top 50 genes ranked by genome-wide expression fidelity.
- CNS region / gene search: The user can search by CNS region / gene to retrieve information about the associations of individual genes with major cell types, as well as top gene expression correlates.
Twenty-one broad CNS regions and the four major CNS cell types (neurons, astrocytes, oligodendrocytes, and microglia) are currently supported. Annotated usage examples are provided below. For additional details, please refer to the publication that accompanies this web site (citation above).
CNS region / cell type search
Fig. 2 provides an example of the ‘wheel plot’ that is returned after querying a particular CNS region / cell type. In this case, the CNS region is ‘All’ and the cell type is ‘astrocyte’. The meaning of each track is explained below the figure caption.
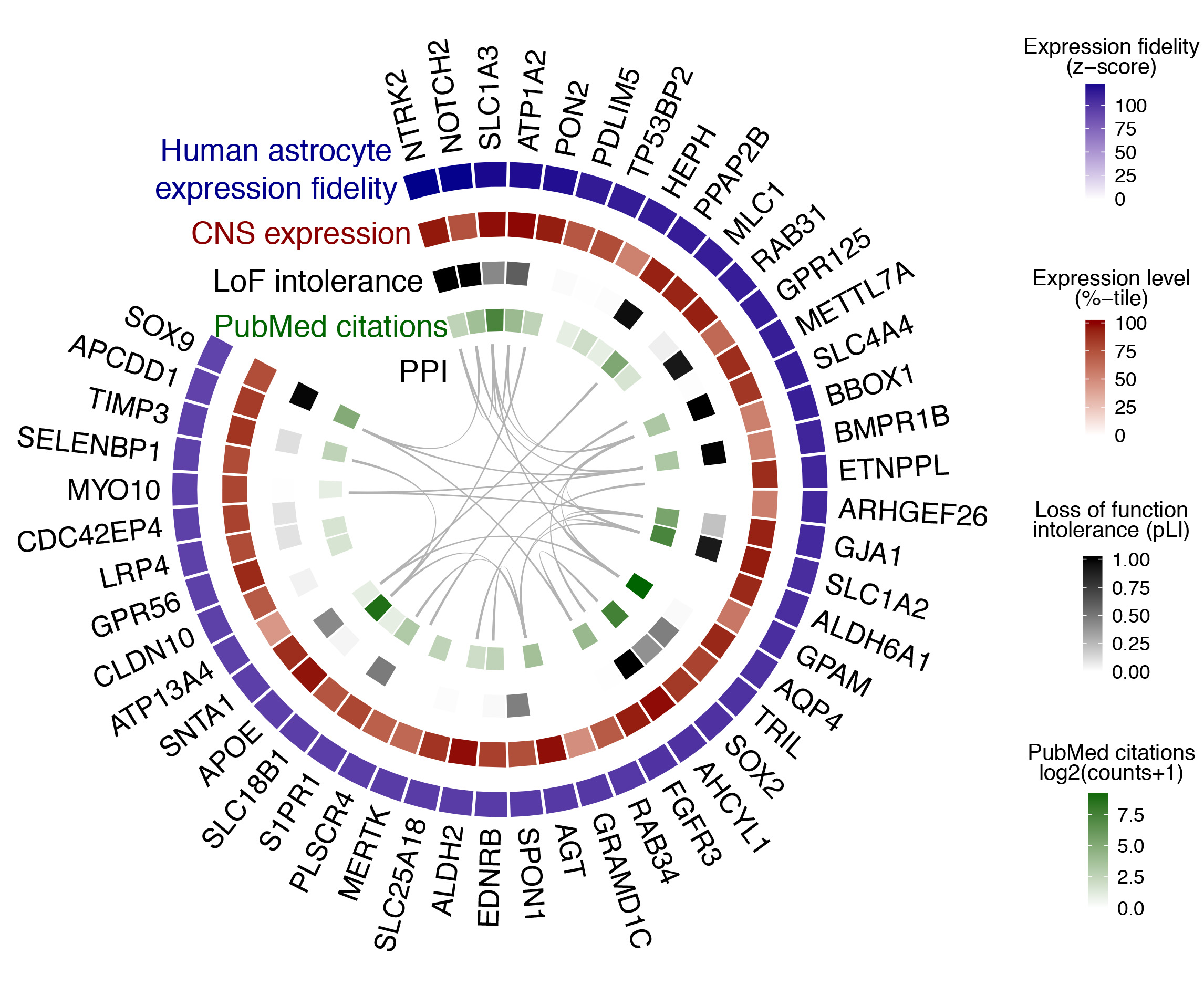
Fig. 2 | Cell type search example. The top 50 genes ranked by expression fidelity for astrocytes in the human CNS (outside track), along with accompanying information for these genes (inside tracks).
- Expression fidelity (blue track) is a z-score that quantifies the extent to which the expression pattern of each gene was correlated with the inferred abundance of the cell type over all queried samples (in this example 7221 human CNS samples were included).
- CNS expression (red track) is an average of the expression percentile ranks of each gene over all queried CNS regional datasets (in this example 62 datasets were included).
- LoF intolerance (black track) is the probability that a given gene is intolerant to loss-of-function (LoF) mutations. Data are from the Exome Aggregation Consortium (ExAC).
- PubMed citations (green track) is the number of citations in PubMed returned by queries for each gene symbol plus the name of the cell type (here, ‘astrocyte’).
- PPI (interior lines) denote protein-protein interactions (PPI) from the STRING database.
Wheel plots are downloadable as PDFs. A complete list of genes ranked by expression fidelity for a given CNS region / cell type, along with absolute expression levels, can be downloaded as a CSV file from the link adjacent to the wheel plot. Complete tables for all CNS regions / cell types are provided on the Data Download page.
CNS region / gene search
Fig. 3 provides an example of the data that are returned after querying a particular CNS region / gene. In this case, the CNS region is ‘All’ and the gene symbol is ‘CNP’. The meaning of each panel is explained below the figure caption.
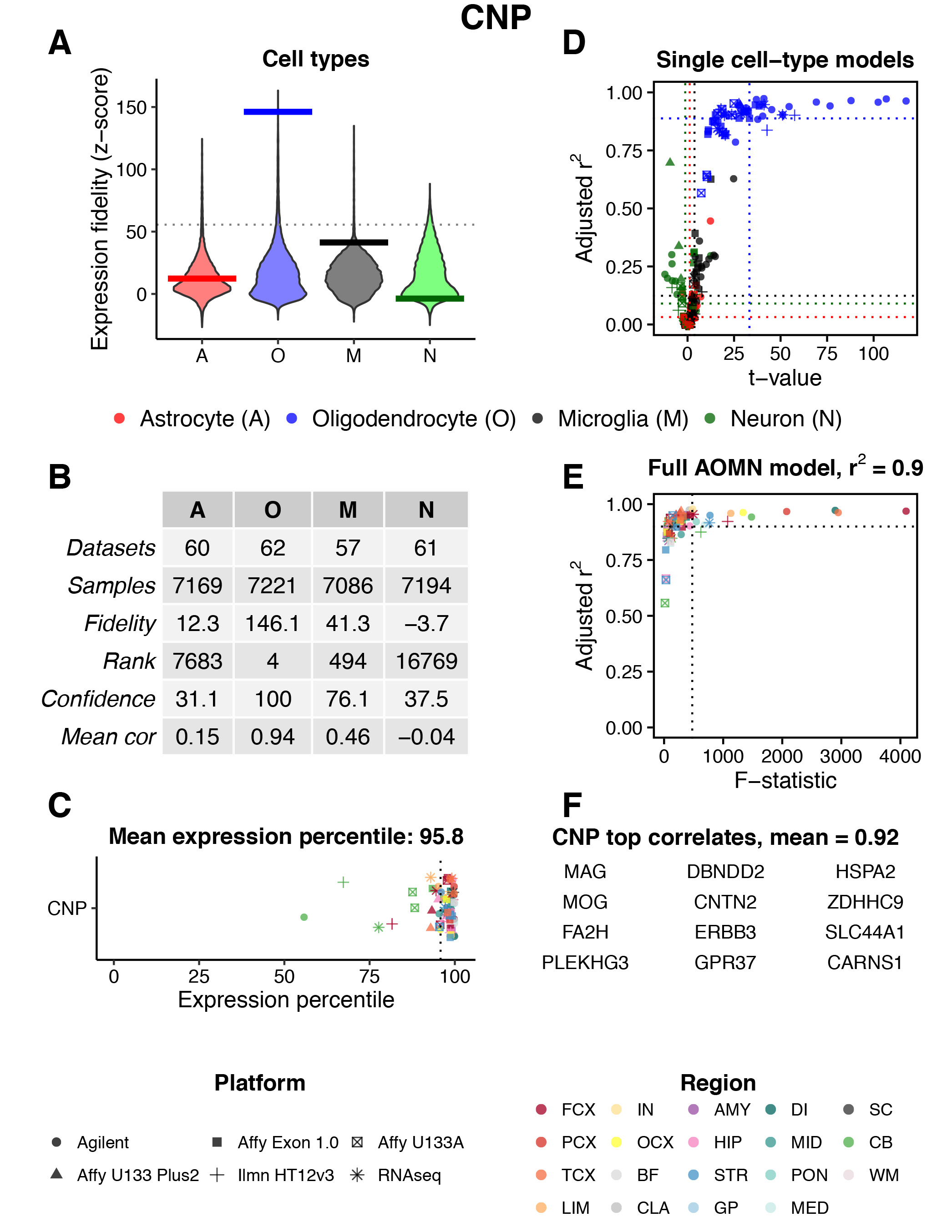
Fig. 3 | Gene search example. Results are shown for CNS region = ‘All’ and Gene = ‘CNP’. The meaning of each panel is described below.
A) Genome-wide distributions of expression fidelity for astrocytes (A), oligodendrocytes (O), microglia (M), and neurons (N) over all analyzed samples are shown. The horizontal line denotes the expression fidelity of the query gene (here, CNP) for each cell type. The dashed horizontal line (only visible when analyzing genes over ‘All’ regions) denotes the threshold above which all fidelity scores had Confidence = 100 (see panel B).
B) This table provides more information on the data used to produce the distributions in panel (A):
- Datasets: the total number of human regional CNS datasets that contributed to the expression fidelity estimates for each cell type.
- Samples: the total number of human CNS samples that contributed to the expression fidelity estimates for each cell type.
- Fidelity: a z-score that quantifies the extent to which the expression of the query gene was correlated with the inferred abundance of each cell type over all analyzed samples.
- Rank: the genome-wide rank of the expression fidelity z-score. Currently the database contains information for ~18,500 genes.
- Confidence: the percentage of genes with expression fidelity scores higher than the query gene that have the highest expression fidelity for that cell type. For example, a confidence score of 100 for oligodendrocytes indicates that the 3 genes with expression fidelity scores for oligodendrocytes that are higher than CNP all show higher expression fidelity for oligodendrocytes than any other cell type.
- Mean cor: the estimated aggregate Pearson correlation coefficient between the expression pattern of the query gene and the inferred abundance of each cell type over all analyzed samples.
C) Mean expression percentile ranks of the query gene in all analyzed datasets. Shapes and colors of points denote technology platforms and CNS regions, respectively.
D) Modeling results for expression levels of the query gene as a function of variation in the abundance of individual cell types. Simple linear regression is used to predict gene expression levels as a function of estimated neuron, astrocyte, oligodendrocyte, or microglia abundance in all human CNS regional datasets containing the query gene. Adjusted r2 values and t-values are shown for each cell type (labeled by color, with shapes denoting technology platforms). In this example, CNP expression is consistently well-modeled by variation in oligodendrocyte abundance across datasets.
E) Modeling results for expression levels of the query gene as a function of variation in the abundance of all cell types. Multiple linear regression is used to predict gene expression levels as a function of estimated neuron, astrocyte, oligodendrocyte, and microglia abundance in all human CNS regional datasets containing the query gene. Adjusted r2 values and F-statistics are shown (with colors and shapes denoting CNS regions and technology platforms, respectively). In this example, the full ‘AOMN model’ can explain ~90% of expression variation for CNP, on average, over all datasets.
F) The top 12 genes ranked by their aggregate correlation to the query gene’s expression levels over all analyzed samples. Among all ~18,500 genes in the database, CNP is most strongly correlated with MAG, followed by MOG, etc.
Region Abbreviations: FCX = frontal cortex; PCX = parietal cortex; TCX = temporal cortex; LIM = limbic cortex; IN = insular cortex; OCX = occipital cortex; BF = basal forebrain; CLA = claustrum; AMY = amygdala; HIP = hippocampus; STR = striatum; GP = globus pallidus; DI = diencephalon; MID = midbrain; PON = pons; MED = medulla; SC = spinal cord; CB = cerebellum; WM = white matter.
Caveats
The data presented on this web site are derived from analyses that are ultimately correlative in nature. Therefore, it is important to acknowledge that covariation of gene expression levels and cellular abundance in bulk tissue samples can be confounded by diverse sources of biological and technical variation. For example, the analyses presented in Fig. 3 indicate that CNP has extremely high fidelity for oligodendrocytes, which is consistent with its reputation as a canonical marker of this cell type. However, CNP also has fidelity for microglia that appears much higher than it does for neurons or astrocytes. This result is driven by covariation of microglial and oligodendroglial abundance in human CNS samples, since both cell types are more abundant in white matter than gray matter. Therefore, variation in the ratio of gray matter to white matter will drive covariation in the abundance of these cell types and the genes that they express. Spurious correlations may also result from alternative splicing, microarray probe failures, batch effects, and limitations in dynamic range (sensitivity / saturation for microarray probes and transcript coverage / read depth for RNAseq). Notwithstanding these limitations, the genes with the highest fidelity for a cell type are already remarkably stable. Ongoing efforts to assimilate new datasets will provide further improvements.